Coronavirus SARS-CoV-2 (formerly known as Wuhan coronavirus and 2019-nCoV) – what we can find out on a structural bioinformatics level
Christian C. Gruber*, Georg Steinkellner
Innophore, Am Eisernen Tor 3, 8010 Graz, Austria
* chirstian.gruber@innophore.com
DISCLAIMER: Permission is hereby granted, free of charge, to any person in academic and/or industrial research and science (independent of their field of interest, country of origin, ethnic group or political system) to obtain a copy of this information and all associated documentation, files, models and data, to deal with the information without restriction (that is to say with no limits to the rights to use, copy, modify, merge, publish, distribute and sublicense copies of the information) and to permit persons to whom the information is furnished to do so, subject to the following conditions: The above copyright notice and this permission notice shall be included in all copies of substantial portions of the information. THE INFORMATION IS PROVIDED “AS IS”, NOT PEER-REVIEWED, WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED, INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE INFORMATION. PARTS OF BACKGROUND INFORMATION SECTION ARE DERIVED FROM PUBLIC SOURCES.
January, 23rd 2020 19:31UTC: 2019-nCoV Background
As of January, 23rd 2020, the Wuhan coronavirus (WHO 2019-nCoV)[i], a positive-sense, single-stranded RNA coronavirus first reported in 2019 is spreading from Wuhan, China, the primary location outbreak. The Chinese government placed the cities of Wuhan, Huanggang, and Ezhou with a combined population of approximately 15 million people, under lockdown in an attempt to contain the viral outbreak e.g. [ii],[iii]. The human-to-human transmission was confirmed in Guangdong, China, according to Zhong Nanshan, head of the health commission team investigating the outbreak.[iv] No specific treatment for the new virus is currently available, but existing anti-virals might be repurposed[v].
According to Wikipedia[vi], sequences of Wuhan betacoronavirus show similarities to beta coronaviruses found in bats; however, the virus is genetically distinct from other coronaviruses such as Severe acute respiratory syndrome-related coronavirus (SARS) and the Middle East respiratory syndrome-related coronavirus (MERS). Like SARS-CoV, it is a member of Beta-CoV lineage B (i. e. subgenus Sarbecovirus). Eighteen genomes of the novel coronavirus have been isolated and reported including BetaCoV/Wuhan/IVDC-HB-01/2019, BetaCoV/Wuhan/IVDC-HB-04/2020, BetaCoV/Wuhan/IVDC-HB-05/2019, BetaCoV/Wuhan/WIV04/2019, and BetaCoV/Wuhan/IPBCAMS-WH-01/2019 from the China CDC, Institute of Pathogen Biology, and Wuhan Jinyintan Hospital. Its RNA sequence is approximately 30 kb in length.
The new genome has led to several protein modeling experiments on the receptor-binding protein (RBD) of the nCoV spike (S) protein. Two Chinese groups, as of 23rd January 2020, believe that the S protein retains sufficient affinity to the SARS receptor (angiotensin-converting enzyme 2, ACE2) to use it as a mechanism of cell entry.
The RNA genome is replicated and a long polyprotein is formed, where all of the proteins are attached. Coronaviruses have a non-structural protein – a protease – which is able to separate the proteins in the chain. This is a form of genetic economy for the virus, allowing it to encode the greatest number of genes in a small number of nucleotides.[vii]
University of Hong Kong School of medicine, has previously said that SARS has been studied earlier and found that protease inhibitors and other drugs can effectively treat respiratory diseases such as SARS, middle respiratory syndrome and other coronaviruses.
There were six kinds of coronaviruses that could infect humans, as well as 24 other kinds that could infect animals including bats, birds, rats, and cows. As most Wuhan patients had connections with the Huanan Seafood Market, there was a high chance the unknown coronavirus was transmitted to wild animals from bats and became mutated before it spread to humans, he said. Usually, a new disease would not be highly infectious between humans so only people who had very close contact with the patients could be infected, he said. If the Wuhan disease was similar to SARS, patients could be potentially cured by doses of ribavirin, protease inhibitor, and interferon.[viii]
Michael Mina, an epidemiologist at the Harvard School of Public Health yesterday said he has heard that some patients in China are being treated with protease inhibitors, antivirals that were developed to treat people with HIV and that were used “somewhat successfully” to treat SARS[ix].
Discovering the protease of coronavirus 2019-nCoV
Innophore decided to allocate significant human- and computational resources to support modelling efforts in this situation. Although Innophore is not active in the field of global epidemics, the fundamental principles of structural enzymology, our main expertise, are independent of the field of application. In the last years we had the chance to work with numerous academic- and corporate partners in the chemical, pharma, nutrition and agricultural industry on many different enzyme classes involving proteases. We worked on proteases used in consumer products, proteases for biocatalytic applications and human and simian IV proteases, studying the interaction dynamics, especially HIV1-protease[x] with various stereoisomers of classical inhibitors like Mozenavir.
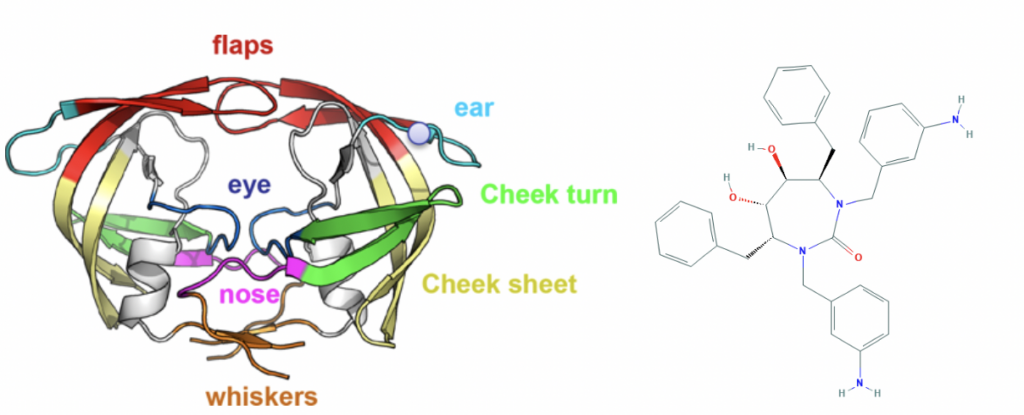
Left: Topology of the HIV-1 protease[xi], Right: HIV-1 protease inhibitor DMP-450/Mozenavir[xii]
Validating the 2019-nCoV protease sequence
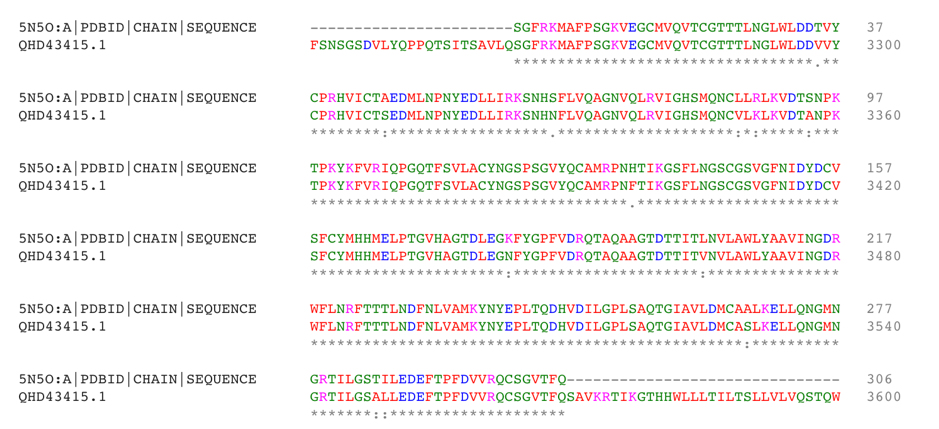
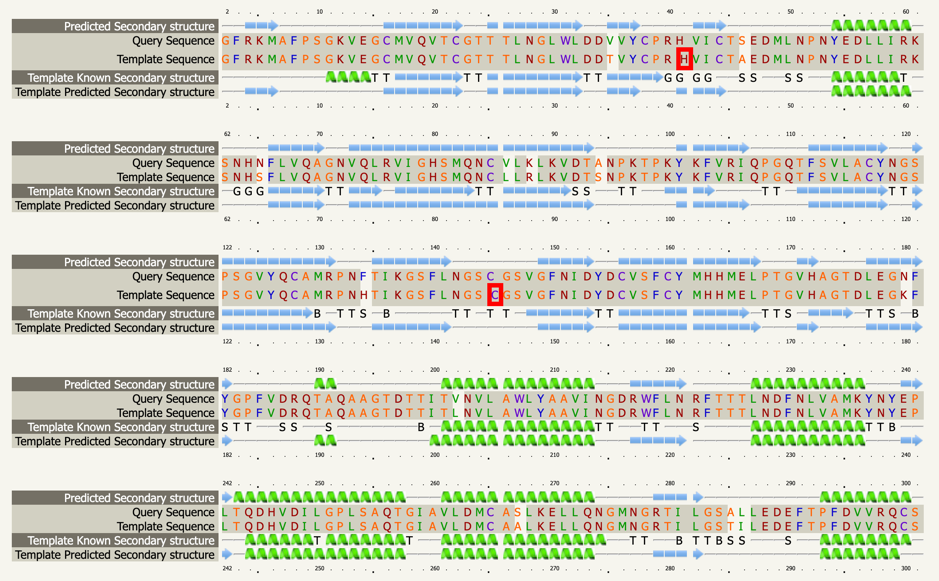
Although there are already modeling activities targeting this virus e.g. [xiii],[xiv], we decided to start from scratch to circumvent any potential biases and to focus on the protein class that our team is most familiar with: The viral protease of 2019-nCoV. Andrew Mesecar, Purdue’s Walther Professor in Cancer Structural Biology and head of the Department of Biochemistry is also working on structure prediction of this target enzyme and the interaction with potential inhibitors. We are waiting for these structures to become publically available. In the meantime, analyzing the viral Wuhan seafood market pneumonia virus genome (NCBI genome ID MN908947[xv], GenBank: MN908947.3) published by Wu, F. Et al. today (LOCUS MN908947, 29903 bp, ss-RNA linear VRL 23-JAN-2020) we identified the potential protease sequence based on multiple sequences alignments with known SARS coronavirus proteases. The following figure shows the aligning sequence region of 2019-nCoV, “orf1ab polyprotein” with protein id QHD43415.1[xvi] with the sequence of PDB entry 5N5O[xvii], a structure of SARS coronavirus main protease deposited by Zhang, L., and Hilgenfeld, R. from the German Center for Infection Research in 2017 using Clustal O:
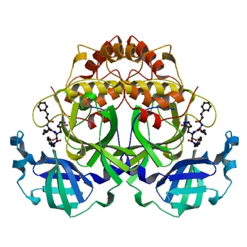 PDB entry 5N5O
PDB entry 5N5O
Using EMBOSS Needle aligning the sequence the translated 2019-nCoV genome with another PDB entry 3TLO[xviii], a crystal structure of HCoV-NL63 3C-like protease, we get the same aligning region:
# Length: 7097 # Identity: 136/7097 ( 1.9%) # Similarity: 192/7097 ( 2.7%) # Gaps: 6795/7097 (95.7%) # Score: 651.5

Extracting the putative protease sequence from position X to Y yields a putative protease sequence of 306 amino acids with a calculated protein weight of 33.8 kilodaltons, which is at the upper range of typical proteases.
>QHD43415.1 putative protease by 5N5O:A sequence alignment SGFRKMAFPSGKVEGCMVQVTCGTTTLNGLWLDDVVYCPRHVICTSEDMLNPNYEDLLIRKSNHNFLVQ AGNVQLRVIGHSMQNCVLKLKVDTANPKTPKYKFVRIQPGQTFSVLACYNGSPSGVYQCAMRPNFTIKG SFLNGSCGSVGFNIDYDCVSFCYMHHMELPTGVHAGTDLEGNFYGPFVDRQTAQAAGTDTTITVNVLAW LYAAVINGDRWFLNRFTTTLNDFNLVAMKYNYEPLTQDHVDILGPLSAQTGIAVLDMCASLKELLQNGM NGRTILGSALLEDEFTPFDVVRQCSGVTFQ
Blasting this sequence again against the PDB revealed proteins with very high sequence similarity and sufficient resolution for subsequent homology modeling, e.g. PDB entry 2A5K[xix].
PDB Sequence Search: SGFRKMAFPS GKVEGCMVQV TCGTTTLNGL WLDDVVYCPR HVICTSEDML NPNYEDLLIR KSNHNFLVQA GNVQLRVIGH SMQNCVLKLK VDTANPKTPK YKFVRIQPGQ TFSVLACYNG SPSGVYQCAM RPNFTIKGSF LNGSCGSVGF NIDYDCVSFC YMHHMELPTG VHAGTDLEGN FYGPFVDRQT AQAAGTDTTI TVNVLAWLYA AVINGDRWFL NRFTTTLNDF NLVAMKYNYE PLTQDHVDIL GPLSAQTGIA VLDMCASLKE LLQNGMNGRT ILGSALLEDE FTPFDVVRQC SGVTFQ Expectation Value = 10.0, Sequence Identity = 0%, Search Tool = blast, Mask Low Complexity=yes)

In total, 138 structures were found meeting a generous expectation value of 10:
1LVO,1P9S,1P9U,1Q2W,1UJ1,1UK2,1UK3,1UK4,1WOF,1Z1I,1Z1J,2A5A,2A5I,2A5K,2ALV,2AMD,2AMP,2AMQ, 2BX3,2BX4,2C3S,2D2D,2DUC,2GT7,2GT8,2GTB,2GX4,2GZ7,2GZ8,2GZ9,2H2Z,2HOB,2K7X,2LIZ,2OP9,2PWX, 2Q6D,2Q6F,2Q6G,2QC2,2QCY,2QIQ,2V6N,2VJ1,2YNA,2YNB,2Z3C,2Z3D,2Z3E,2Z94,2Z9G,2Z9J,2Z9K,2Z9L, 2ZU2,2ZU4,2ZU5,3ATW,3AVZ,3AW0,3AW1,3D23,3D62,3E91,3EA7,3EA8,3EA9,3EAJ,3EBN,3F9E,3F9F,3F9G, 3F9H,3FZD,3IWM,3J1Z,3M3S,3M3T,3M3V,3MOG,3SN8,3SNA,3SNB,3SNC,3SND,3SNE,3SZN,3TIT,3TIU,3TLO, 3TNS,3TNT,3V3M,3VB3,3VB4,3VB5,3VB6,3VB7,4F49,4HI3,4MDS,4RSP,4TWW,4TWY,4WMD,4WME,4WMF,4WY3, 4XFQ,4YLU,4YO9,4YOG,4YOI,4YOJ,4ZRO,4ZUH,5B6O,5C3N,5C5N,5C5O,5EU8,5GWY,5GWZ,5HYO,5N19,5N5O, 5NH0,5NH0,5NH0,5VRF,5WKJ,5WKK,5WKL,5WKM,5ZQG,6FV1,6FV2,6JIJ
For modeling the 3D structure of 2019-nCoV protease, we used our CatalophoreTM platform as well as the public Phyre2[xx] server to generate homology models. Both approaches yielded satisfying results as expected given by the very high sequence similarity.
Phyre2 top model d2duca1 is based on the fold library id d2duca1[xxi], a trypsin-like serine protease oft the viral cysteine protease of trypsin fold from the SARS coronavirus main proteinase[xxii].
 Phyre2 confidence in the model is 100.0%, although we wouldn’t go that far.
Phyre2 confidence in the model is 100.0%, although we wouldn’t go that far.
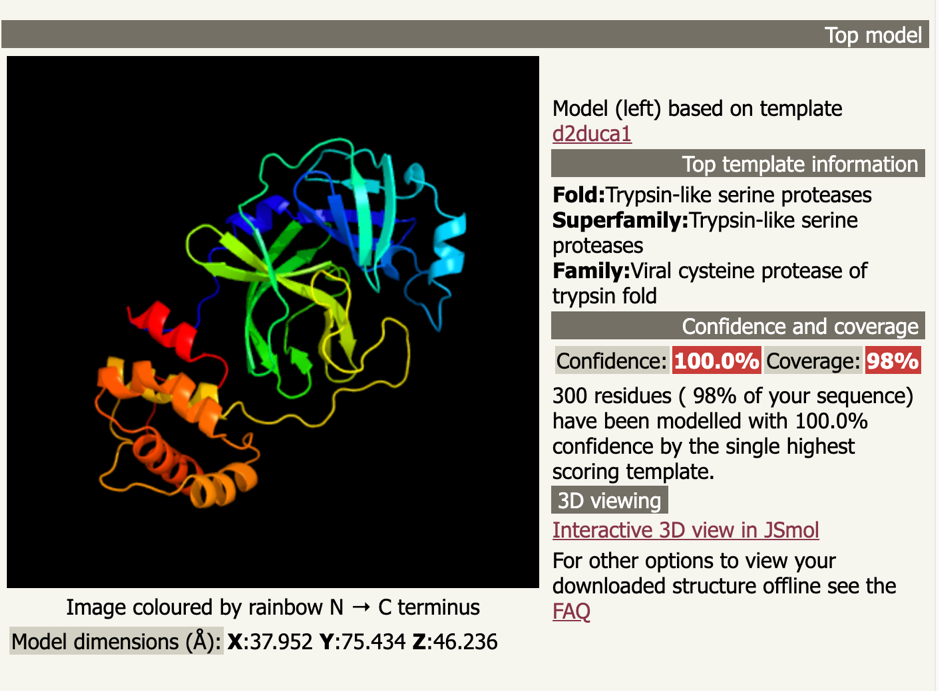
You can download the complete Phyre2 run here: d06ff0dcb8400814.tar
In subsequent steps, we will identify cavities in the homology models, annotate them to generate point clouds.
Innophore’s CatalophoreTM platform predicated a homology model bases on the structural template 2H2Z[xxiii], chain A, the crystal structure of SARS-CoV main protease with authentic N and C-termini, with an overall quality “Pretty good”. We expect the protein to be a monomer. You can download the model as compressed PDB file here: QHD43415_1-putative-protease_cleaned.pdb
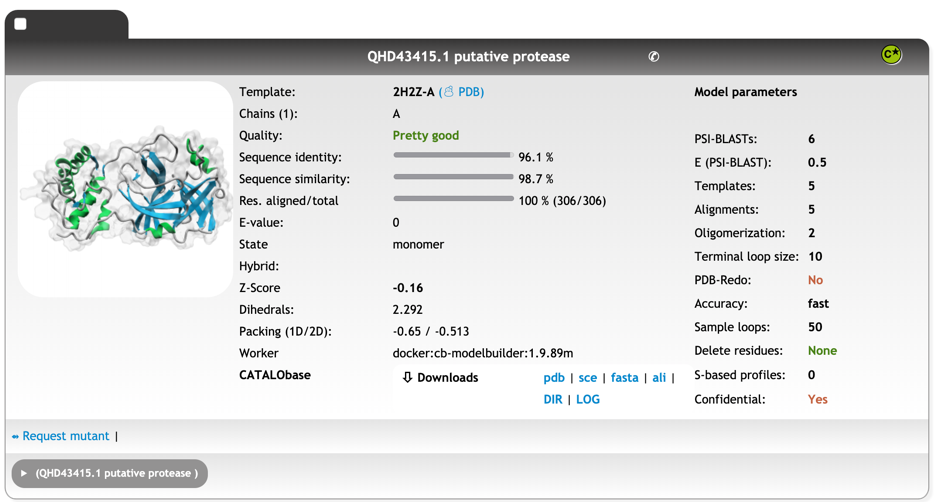
Sequence identity: 96.1 %
Sequence similarity: 98.7 %
Res. aligned/total: 306/306
The comparison of the two independent models showed high similarity, with a final RMSD of 0.396 Å:
Match: read scoring matrix. Match: assigning 300 x 306 pairwise scores. MatchAlign: aligning residues (300 vs 306)... MatchAlign: score 1616.000 ExecutiveAlign: 300 atoms aligned. ExecutiveRMS: 7 atoms rejected during cycle 1 (RMSD=0.89). ExecutiveRMS: 14 atoms rejected during cycle 2 (RMSD=0.62). ExecutiveRMS: 12 atoms rejected during cycle 3 (RMSD=0.54). ExecutiveRMS: 15 atoms rejected during cycle 4 (RMSD=0.49). ExecutiveRMS: 9 atoms rejected during cycle 5 (RMSD=0.43). Executive: RMSD = 0.396 (243 to 243 atoms)
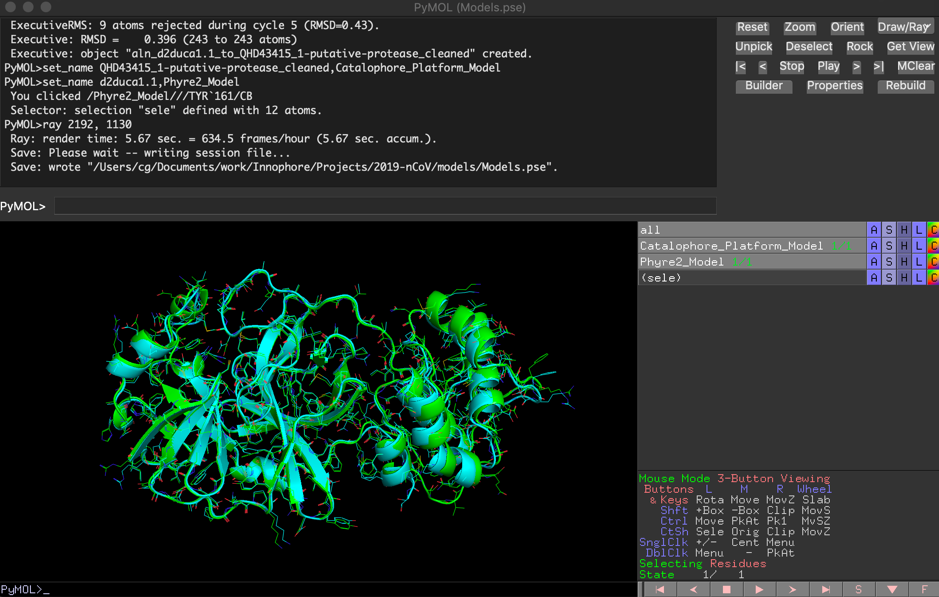
The PyMol session file containing both models can be downloaded here: 2019-nCoV_putative_protease-Models.pse
2019-nCoV active sites considerations
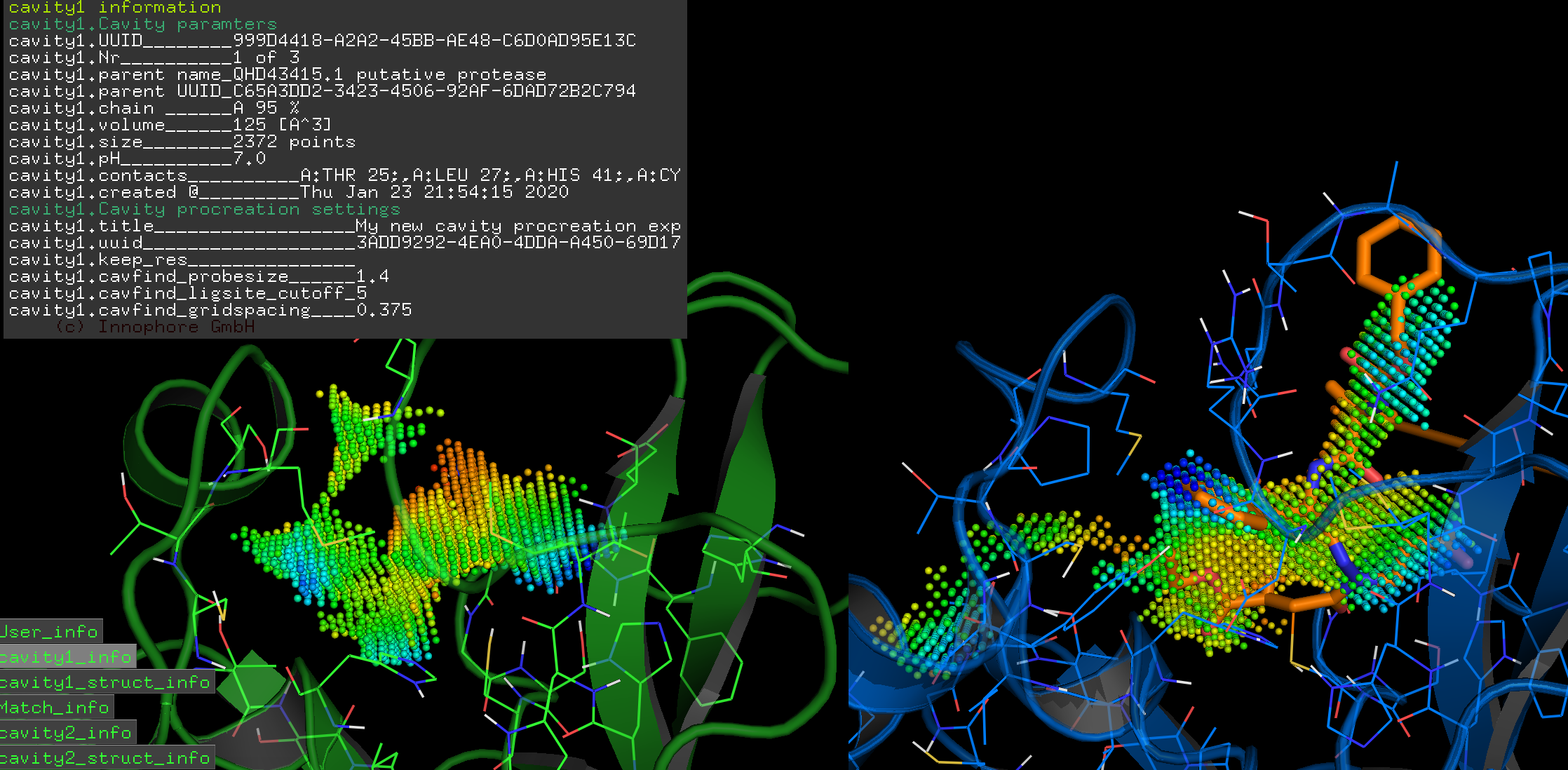
For the template structure 2H2Z, our CatalophoreTM database has point-cloud cavities of 6 cavity breeds available, calculated under different environments (e.g. pH):

At protonation state pH 7, we have two cavities for the template, both annotated as EC 2.7.48:

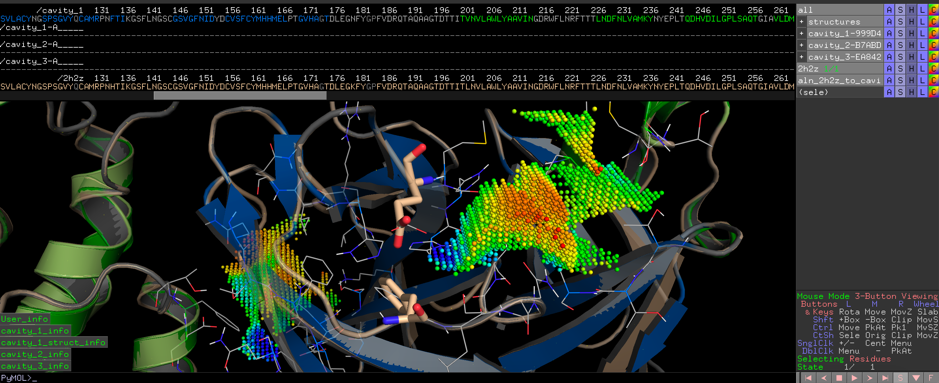
We calculated the active site CatalophoreTM point-cloud for the putative 2019-nCoV protease. Using standard setting, we obtained three cavities in the 2019-nCoV protease model:
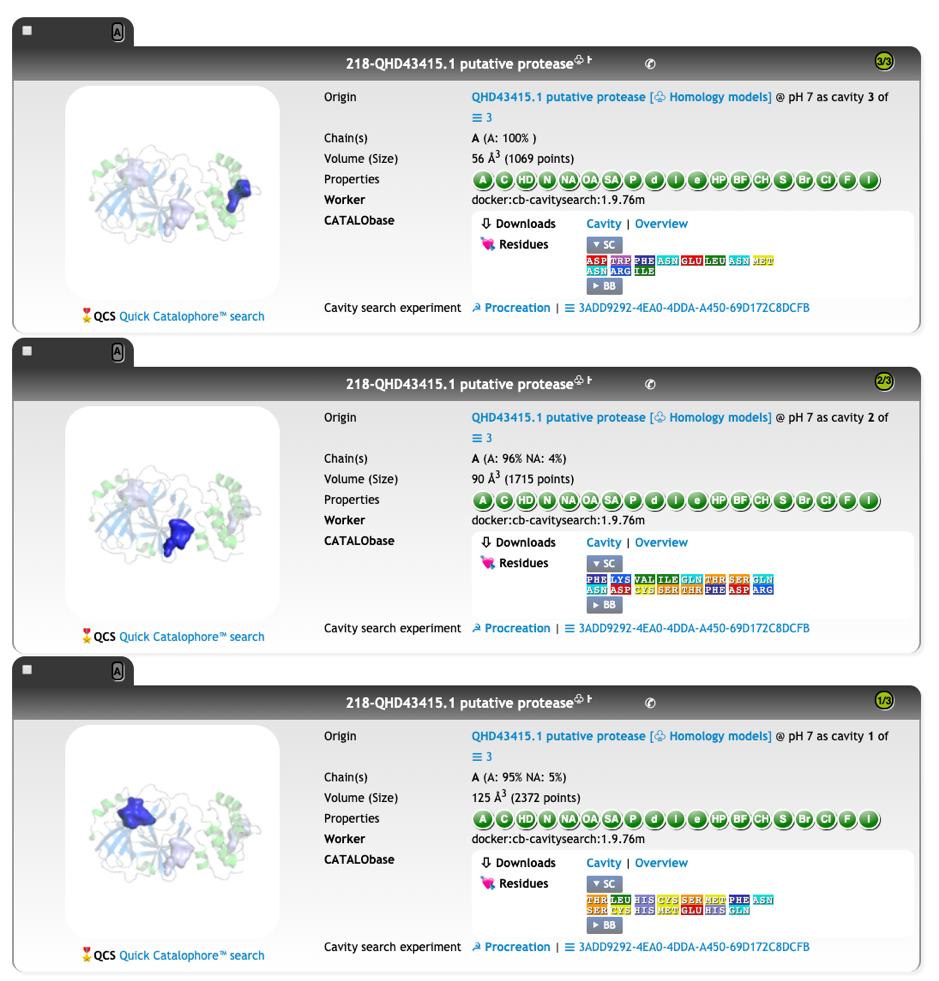
We are currently fingerprinting the most likely candidate for the active site and re-checking our cavity procreation parameters. We will come back shortly with a downloadable version including the physicochemical parameter point-clouds and analysis of the differences to the proteases from other coronaviruses.
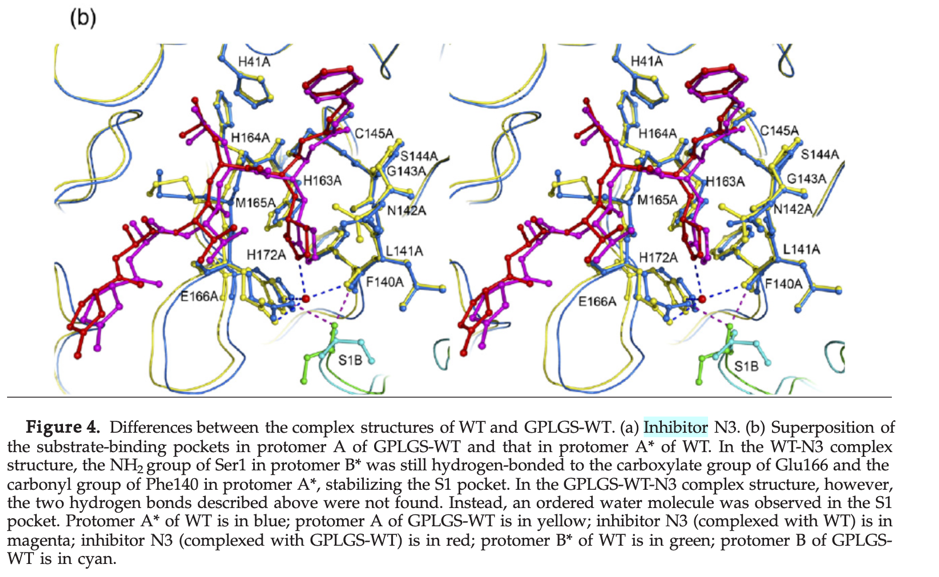
By aligning PDB entry 2H2Z from Yang, H. Et. al 2006[xxiv] with our model and mapping the residues Glu166 and Phe140 (figure above) of the inhibition site to our point-cloud CatalophoresTM sites, we could identify cavity “1” to be a potential target site for inhibition.
Suqsequentially we will search our CatalophoreTM databases for similar point clouds in hope of identifying proteins with similar distribution patterns in the physicochemical property space with known inhibitors to potentially find inhibitors that bind to the protease of the Wuhan virus as well.
Update January, 24rd 2020 1:24 UTC: Catalophore search started
We reallocated 3/4 of our computational resources – several thousand cores – to screen 535.879 cavities derived from the PDB overnight. The estimated total CPU time for this screening is approximately 23 days.
One of the first meaningful matches out of the 15% that were screened until now is a cavity match with a point-cloud from PDB entry 2A5I (https://www.rcsb.org/structure/2A5I, Crystal structures of SARS coronavirus main peptidase inhibited by an aza-peptide epoxide in the space group C2):
match-306-CDDEE285-3E3F-11EA-A0D6-0242C0D00007.zip
Matching results
Match Total score 0.03919460
Match Distance score 0.00555660
Match Overlap 1 79 %
Match Overlap 2 52 %
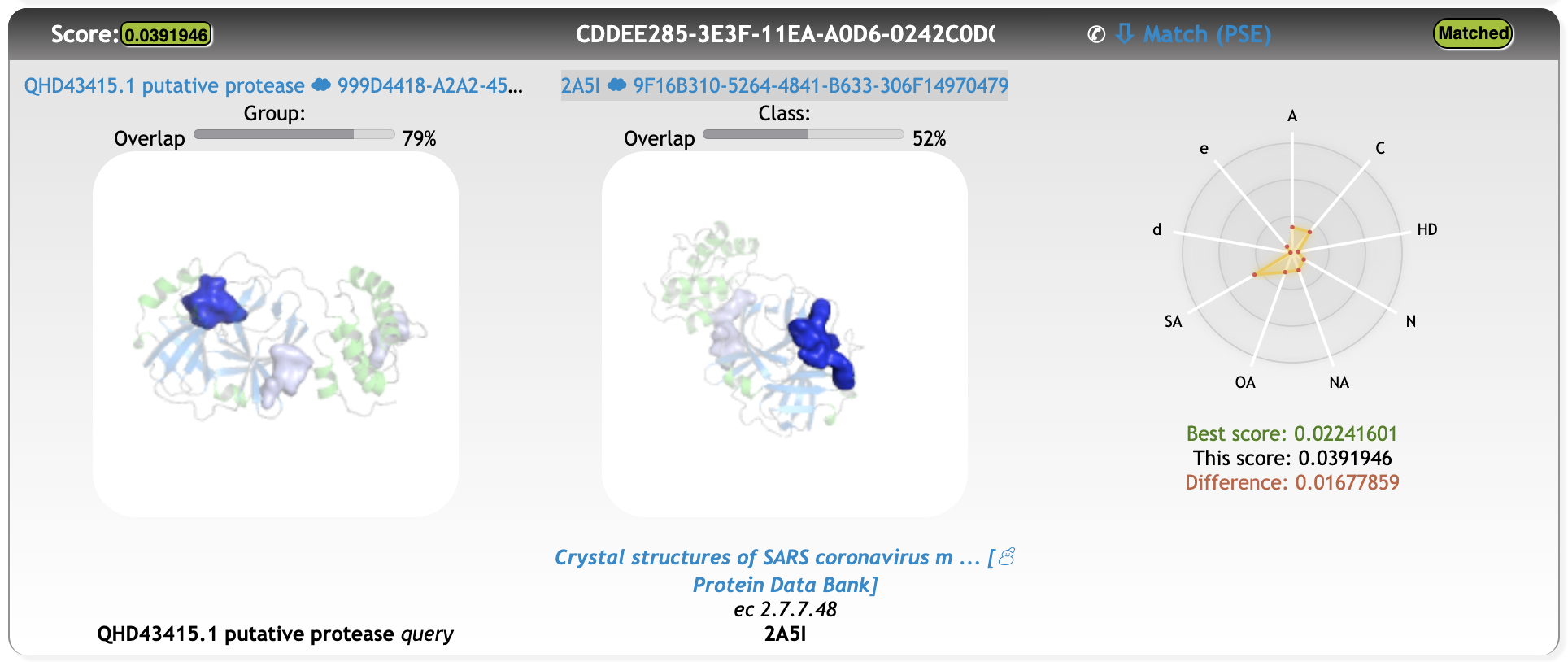
Although the cavity overlap is not perfect, the alignment of the protein structures solely based on the cavity rotation-translation matrix is satisfying.
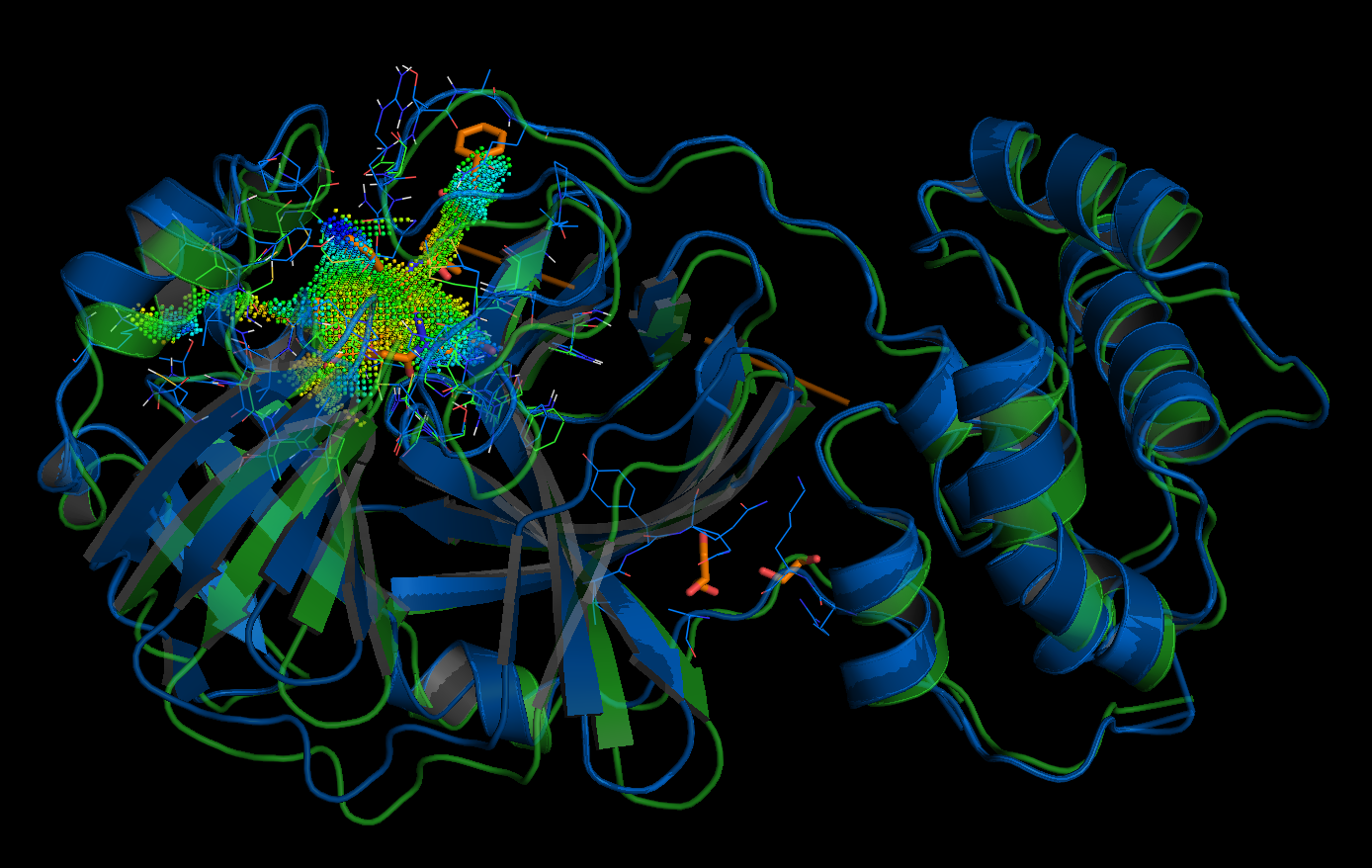
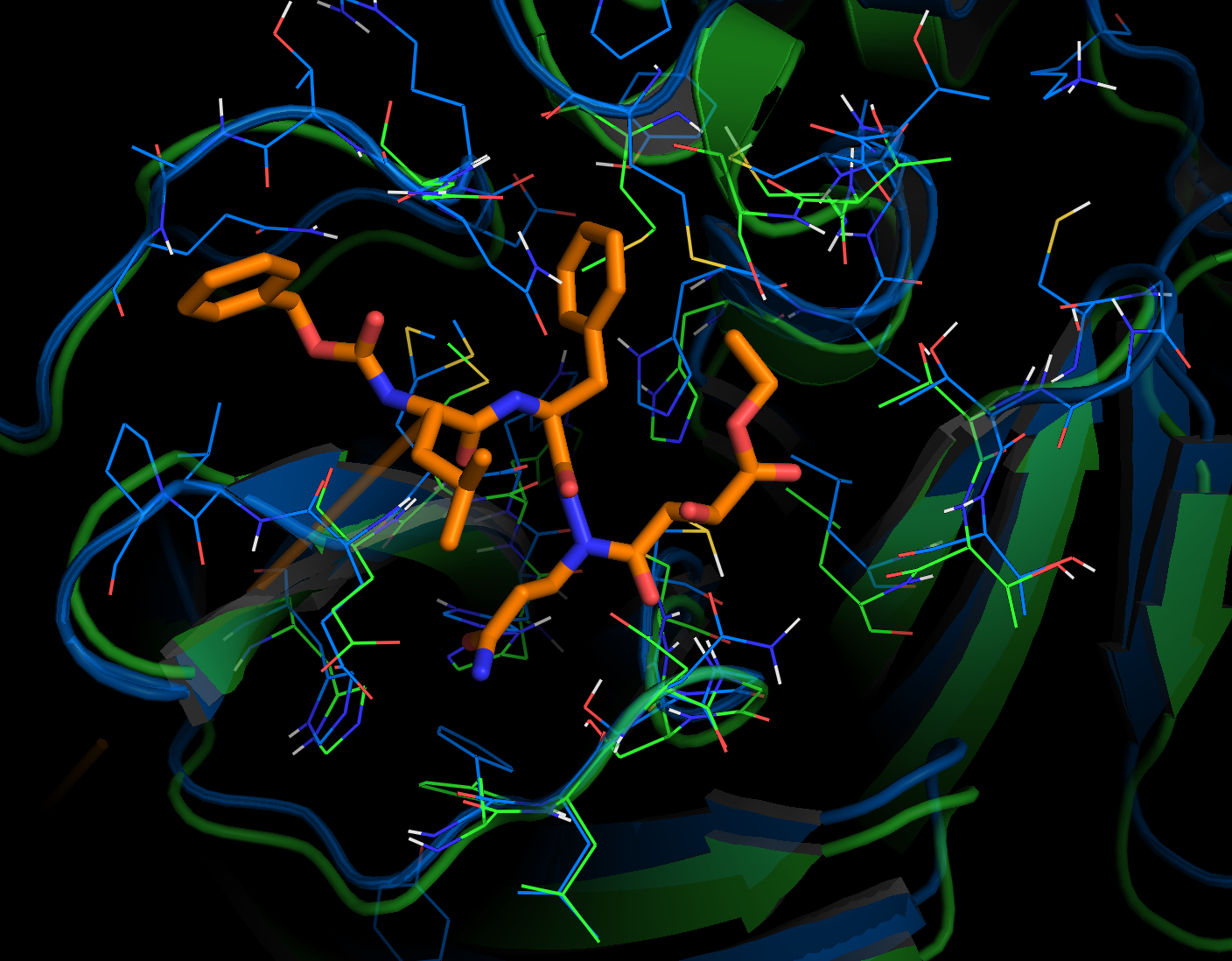
Left: active site of the putative 2019-nCoV protease, right: database entry FDBB501F-1EF4-4AC9-8AA3-0CE76, the point-cloud of the SARS coronavirus peptidase inhibited by an aza-peptide epoxide:
Overlay of the compound AZP (https://www.rcsb.org/ligand/AZP) based on the cavity matching alignment. This is not a docking result – the coordinates of the ligand were transformed based on the cavity match and transferred onto the 2019-nCoV protease.
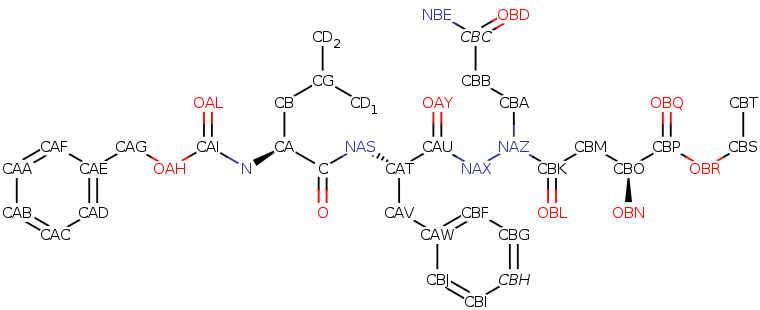
Ethyl (5S,8S,14S)-1
1-(3-amino-3-oxopro pyl)-8-benzyl-14-hy droxy-5-isobutyl-3, 6,9,12-tetraoxo-1-p henyl-2-oxa-4,7,10, 11-tetraazapentadec an-15-oate
Molecular Formula: C32H43N5O9
Average mass: 641.712 Da
Monoisotopic mass: 641.306091 Da
ChemSpider: ID4450034
Update January, 24rd 2020 2:16 UTC: Catalophore search finished
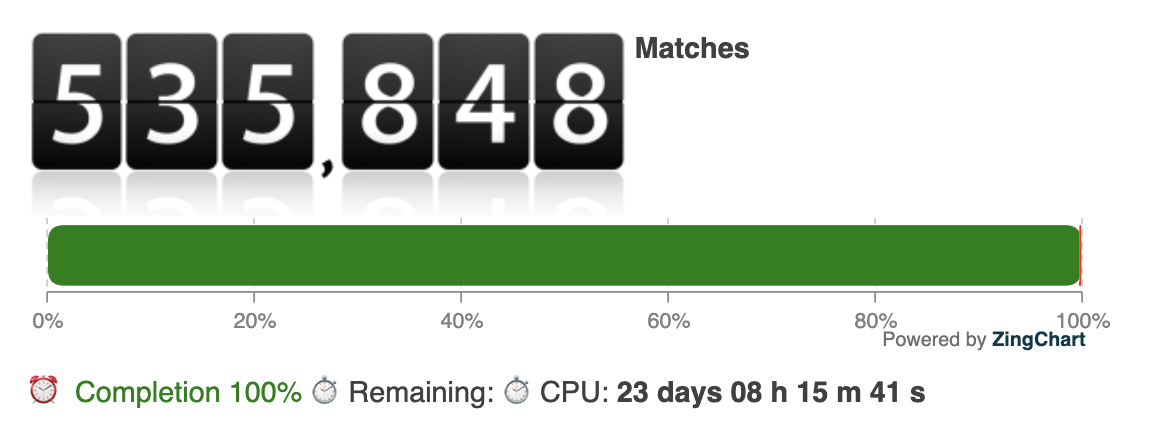
Our in-silico Catalophore screening completed after 01 h 38 m 21 s real-time, roughly the expected 23 CPU days. We now filtered the results for Catalophore point-clouds in the database with an overlap of more than 70% with the 2019-nCoV virus protease point-cloud and favored cavities were crystallographic ligands were bound before calculating the cavities. We limited the results to cavities larger than 150A^3 covering the complex ligand to enrich medium- to large-sized organic compounds in the ranked list.
Disclaimer: This following list does not take into account any pharmacological-, toxic- or side effect nor does it represent compounds directly suggested as potential drugs against 2019-nCoV. The list currently contains 148 organic compounds in total, that bind to protein cavities that share high physicochemical similarity to the 2019-nCoV protease active site cavity based on our multi-dimensional point-cloud matching.
The preliminary top 5 hits potentially binding to the putative 2019-nCoV protease are listed in the following table – after further inspection, the remaining candidates will be available too:
| Catalophore Score | Compound | Formula | InChIKey |
| 0.023 | G75 | 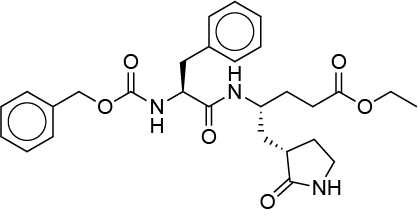 |
PIZHLOUXQJUQHF-VXNXHJTFSA-N |
| 0.026 | G82 | 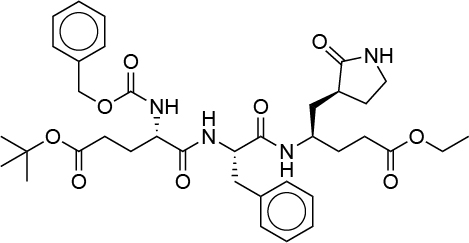 |
MMMLJIROCXIHMV-XJYHXZFBSA-N |
| 0.027 | DTZ | KEQKAMYELZXRRN-UHFFFAOYSA-L | |
| 0.030 | R30 | 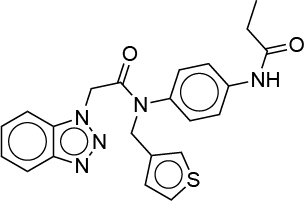 |
TWIVXCFEBRGEKY-UHFFFAOYSA-N |
| 0.031 | 3X5 | 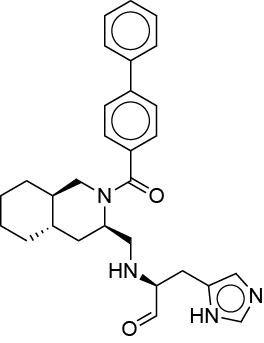 |
VZCULZJNALRGNB-DNZWLJDLSA-N |
Update January 24th 2020, 16:00UTC
Since we were mentioned in the Wikipedia article today about the novel coronavirus 2019-nCoV having published comparative models and preliminary inhibitors of the #2019-nCoV protease we are in contact with several official bodies to further contribute to the field.
Update January 24th 2020, 22:00UTC
Thanks to our colleagues from the GISAID initiative, tonight we have gained access to 17 additional nCoV genome data sets recently derived from patients.
Update January 25th 2020, 00:30UTC
We are so proud to be working with a group of bioinformaticians from a major pharmaceutical company in Beijing and the Chinese CDC since 1 a.m. to search our Catalophore databases for potential experimental or approved drug targets that could bind to the #2019-nCoV protease and to review our data with our colleagues in China.
Update January 25th 2020 09:30UTC
More than 20 research groups incl. the Shanghai Institute of Materia Medica, Chinese Academy of Sciences and Shanghai University of Science and Technology’s Institute of Immunochemistry joined the emergency response team against #2019-nCoV virus infection, using the accumulated anti-SARS drug research experience to conduct anti-2019-nCoV drug research. A list of potential compounds was published just now. The HIV-1 protease inhibitor DMP450 we mentioned yesterday night in our post is not in the list, however many of the entries are HIV inhibitors. We were informed by CDC, that Innophore’s top 2 ranked candidate molecules from yesterday, G75 and G82 are missing drug status. So the search will be focused on approved drugs only now. Still, these molecules are supposed to be the potentially the best binders, derived from the analysis of the previously published crystal complexes e.g. by Lee Et. al (https://www.rcsb.org/structure/2A5I). Molecular dynamics analysis will be available in 4-5 hours. Some of the compounds listed by the emergency response team are found in crystal structures of complexes and are highly ranked in cavities of our yesterday Catalophore search, meaning having a total score under 0.1, e.g. Lopinavir (is the Top3 candidate of the Chinese emergency response team) and scored in our search with a Catalophore total score of 0.085939 by matching our cavity of 2019-nCoV protease cavity with the cavity of PDB 1MUI (https://www.rcsb.org/structure/1MUI, published in 2002). So Lopinavir is potentially one of the better binders to the Wuhan coronavirus 2019-nCoV protease cavity – and a previously approved “old” drug (https://de.wikipedia.org/wiki/Lopinavir).
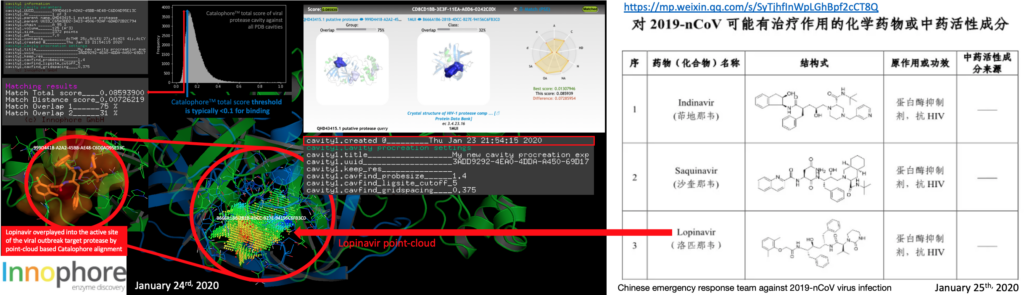 Update January 25th 2020 10:03UTC
Update January 25th 2020 10:03UTC
As mentioned, we are working with a group of bioinformaticians from a major pharmaceutical company in Beijing and the Chinese CDC since 1 a.m. to search our Catalophore databases for potential experimental or approved drug targets that could bind to the #2019-nCoV protease and to review our data with our colleagues in China. We have to coordinate the file transfers and communication with our partners in China, therefore we stop to publish now to get the work done. Cross your fingers and if you have any suggestions, contact us anytime.
Update January 25th 2020 10:25UTC
Since the last days, the requests for our webserver are continuously increasing. Since this night, it’s even growing much faster. Our website is hosted at an external provider – if the server breaks under the load we will migrate to AWS or Google with the following updates – if any. We would post the links on LinkedIn.
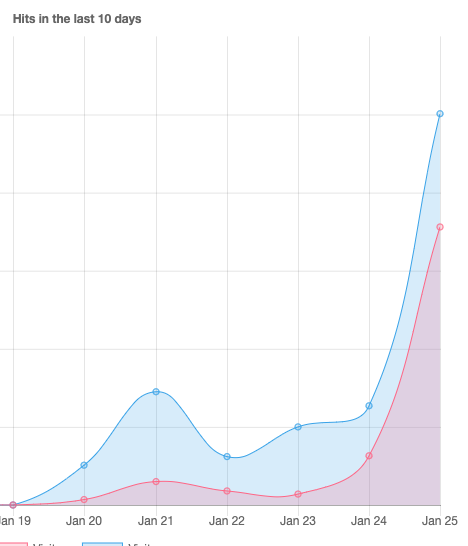
Update January 29th 2020 16:10UTC
We were rather busy since Saturday to confirm, reevaluate, screen and communicate our data about ncov protease. Refering strictly only to public media here, f.i. NYT from yesterday without any other governmental, industrial or scientific or medical source from any continent, it’s officially confirmed since yesterday that China tests Lopinavir for the treatment of the coronavirus in hospitals. Lopinavir was identified as one of the top targets by Innophore Catalophore search last week. This compound has already shown promising results in SARS that has a structurally very similar protease. We finished running the (short) MD simulations on different conformations based on the Catalophore point-cloud alignment and (re)- docking lopinavir into the 2019ncov virus protease model. The docking experiment produced 8 clusters of possible conformations, we chose 6 out of 8 conformers and ran an all-atom 300 ps MD at 310 K (=36.85°C) for each one of them. The conformers have been named LP1 (highest binding energy) to LP6 (lowest binding energy) based on the docking clustering. We will publish the trajectories today here a little later.
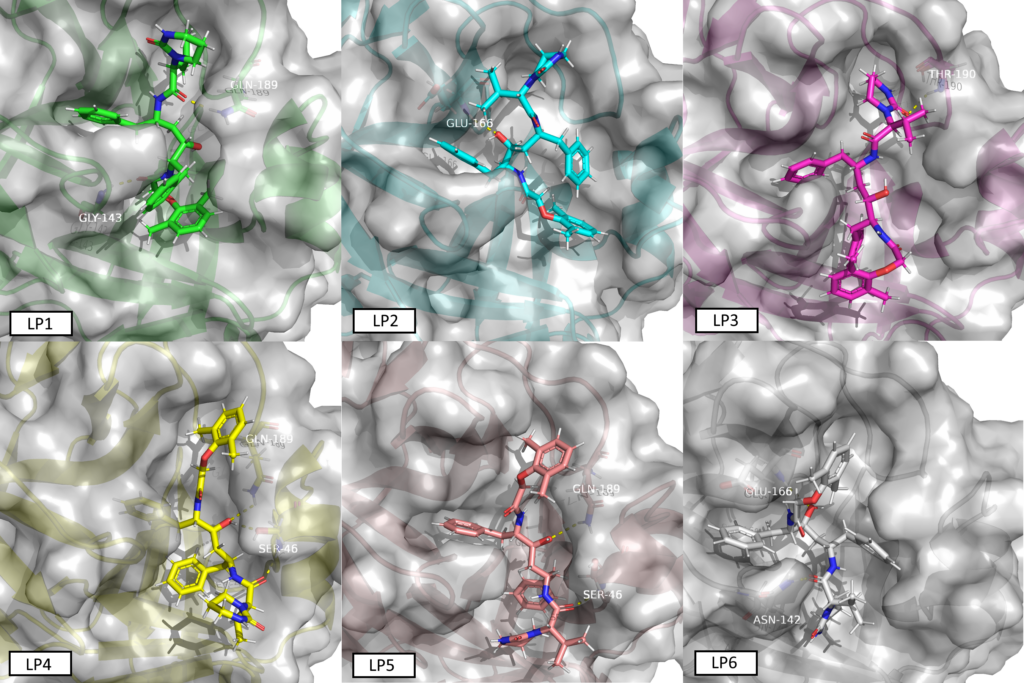
The conformer in the video is the number LP1.
Update January 28th 2020 21:30UTC
A 2019-ncov protease model was published by Xu etl al. yesterday. Their sequence alignment is basically identical to ours from 23rd of January. However, we couldn’t find the structure files for download. If anybody has access to a model that differs significantly from hours, please let us know.
Update January 29th 2020 00:05UTC
We created DOIs for this information, please refer to 10.6084/m9.figshare.11752749 if you want to cite this document or to 10.6084/m9.figshare.11752752 if you want to refer to the model and the renderings in your publications.
Update January 29th 2020 01:10UTC
Standby.
Update January 29th 2020 18:55UTC
Yesterday we started to look for further enzymes in the genome of the 2019-ncov virus. We have no data available until now.
Molecular dynamics simulations of the comparative model of novel coronavirus 2019-nCoV protease Mpro in complex with 6 different conformations based on the Catalophore point-cloud alignment and (re)- docking of lopinavir into the 2019ncov virus protease model are now available for download here 10.6084/m9.figshare.11764158. The initial docking experiment produced 8 clusters of possible conformations, we chose 6 out of 8 conformers and ran an all-atom 300 ps MD at 310 K (=36.85°C).
- The two images in the main folder refer to the docked structures before the MD simulation.
- The file all_centroids.pse contains the frames representing the centroid of the subsequent MD simulation for each docking cluster.
- Each archive contains the centroid in PDB format, the starting frame of the simulation in GRO format and the compressed trajectory in XTC format. In the directory “other_files” there are other data generated during the simulation, i.e. heatmap representing the contact frequency between the ligand atoms and the ones belonging to the homology model.
Update January 29th 2020 21:08UTC
LP1 heatmap representing the contact frequency between the ligand atoms and the ones belonging to the homology model.
![]() Update February 1st 2020 17:01UTC
Update February 1st 2020 17:01UTC
Yesterday (31.1.2020 15:45UTC) we published an analysis (download at 10.6084/m9.figshare.11778750) showing the H-bond network established during the 6 molecular dynamics simulations of coronavirus 2019-nCoV protease model in complex with different conformations of lopinavir (see figure from Update January 29th 2020 16:10UTC above). The dashed lines reported in the said structures do not necessarily reflect the presence of a hydrogen bond in the particular frame displayed (the “centroid” of the simulation), but indicate that the bonds occurred at least once during the simulation. Point clouds generated by the Catalophore platform are represented in sphere mode and their color represents the buriedness index. Point clouds were calculated on the whole active site of the enzyme disregarding the presence of a ligand. A textual list of the H-bond interactions is below. This data could help to design improved variants of the ligand. In the following figure, LP1 is shown. The PyMol download file includes all LPs.
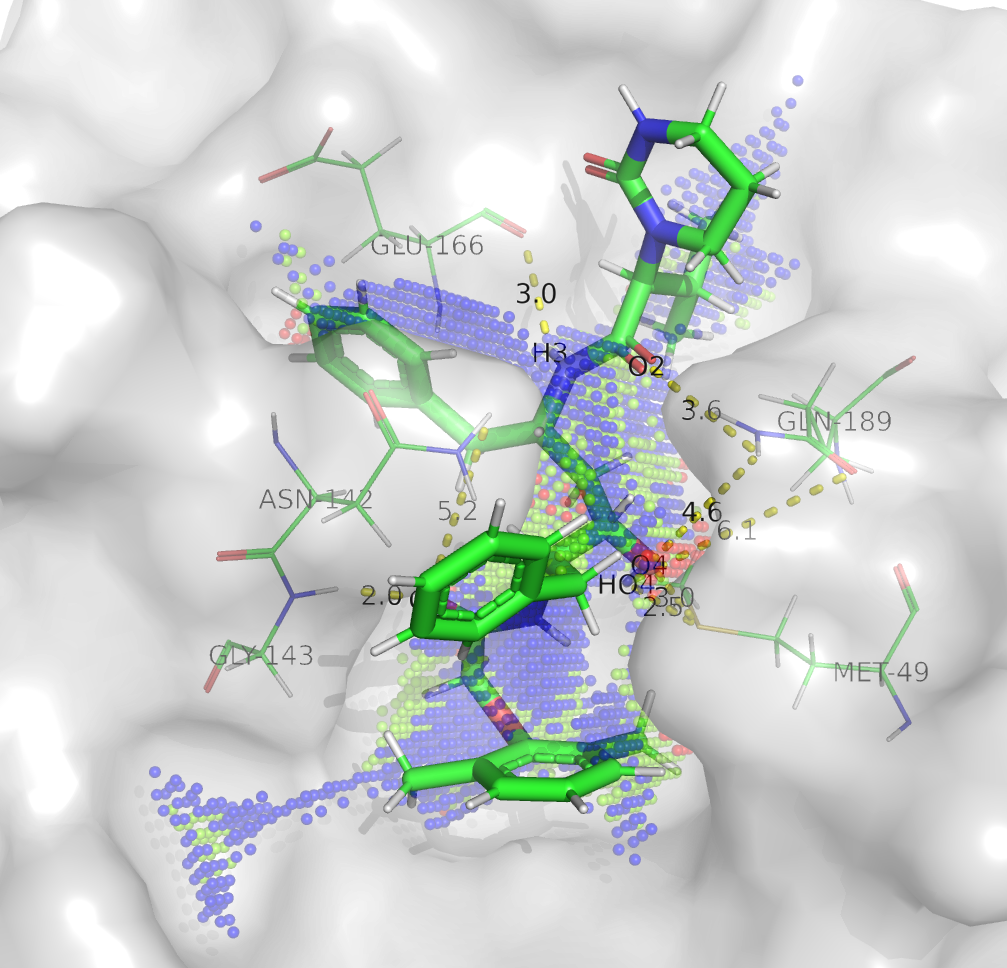
sc: sidechain, bb: backbone
- LP1
Met49 sc: H4-HO4
Asn142 sc: O5
Gly143 bb: O5
Glu166 bb: H3
Gln189 sc: O4-HO4-O2 - LP2
Asn142 sc: O5
Glu166 bb: O4
Gln189 sc: O2 - LP3
Ser46 sc: O3-O5
Asn142 sc: O4
Met165 sc: HN2
Gln189 sc: O1-H3
Thr190 bb: O1-HN2 - LP4
Thr24 sc: O1
Ser46 sc: O1-O2-O4-HO4
Met165 sc: H4
Gln189 sc: O4 - LP5
Thr24 bb: HN2
Ser46 bb/sc: bb: O2; sc: O4-HO4
Met165 sc: H4
Gln189 sc: H4-O4-HO4-O5 - LP6
Thr26 bb: HN2
Asn142 sc: O2-O5
Gly143 bb: O2
Cys145 sc: HO4
His163 sc: HO4
His164 sc: HO4
Met165 sc: HO4
Glu166 bb/sc: bb: H4-O4-HO4; sc: H4
Side note: We double checked our and the other groups alignments of the protease and did not identify representative insertions that are found in other species.
Comparison to other 2019-ncov homology models
In the meantime, additional models were built, the RMSD to our published models are in the range of 0.3 and 0.9A. The Zhang lab yesterday published on their website models for all protein products of 2019-ncov virus (https://zhanglab.ccmb.med.umich.edu/C-I-TASSER/2019-nCov/). The mPro protease we are dealing with is called “QHD43415_5 (L=306)” on their website. The alignment yields an RMSD of 0.9A:
Match: read scoring matrix.
Match: assigning 306 x 306 pairwise scores.
MatchAlign: aligning residues (306 vs 306)…
MatchAlign: score 1646.000
ExecutiveAlign: 306 atoms aligned.
ExecutiveRMS: 3 atoms rejected during cycle 1 (RMSD=1.47).
ExecutiveRMS: 5 atoms rejected during cycle 2 (RMSD=1.00).
ExecutiveRMS: 4 atoms rejected during cycle 3 (RMSD=0.95).
ExecutiveRMS: 3 atoms rejected during cycle 4 (RMSD=0.92).
ExecutiveRMS: 4 atoms rejected during cycle 5 (RMSD=0.91).
Executive: RMSD = 0.886 (287 to 287 atoms)
Executive: object “aln_QHD43415_5_to_Catalophore_Platform_Model” created.
Green: Catalophore Platform, cyan: Phyre2, yellow: I-TASSER, rose: SwissModel
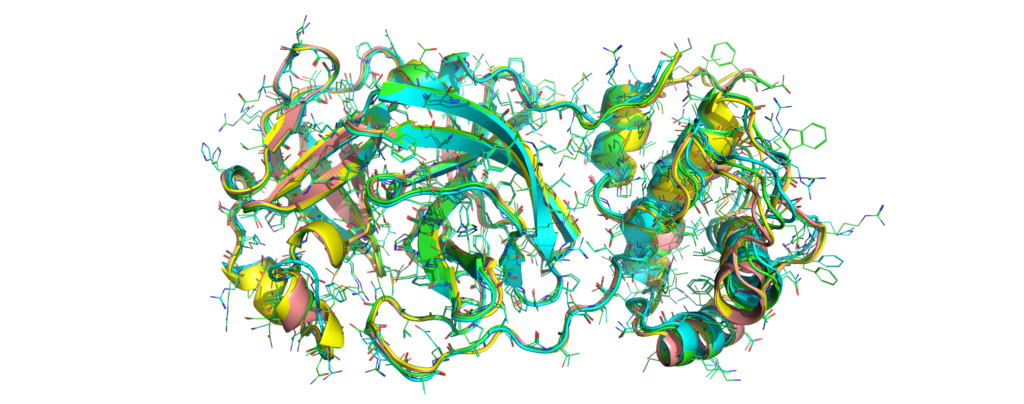
The active site constellations are close. Still, we will publish a detailed cavity comparison of the available models in the next days.
Update February 2nd 2020 21:05UTC
Our partners Horacio Pérez-Sánchez, José Pedro Cerón-Carrasco, Jorge Peña-García and Antonio Jesús Banegas-Luna from the Structural Bioinformatics and High Performance Computing Research Group (BIO-HPC, http://bio-hpc.eu) from Universidad Católica de Murcia (UCAM, http://www.ucam.edu) used SBVS and LBVS methods to screen for potential inhibitors and sent us the results yesterday to publish on our channels.
A) first list (SBVS), first we used our Blind Docking approach. We wanted to check if a blind redocking calculation would allow the co-crystallized ligand when put out apart from the protein to 1) find the active site, and 2) align it to the crystal pose with less than 2A RMSD. Our BD method is reported in https://www.nature.com/articles/s41589-019-0278-6.We tried several docking engines at the same time underneath the BD approach; Autodock Vina, Autodock 4, Lead Finder, FRED (Open Eyes). And we found FRED could do it and LF was rather close. So from then one we just used those two methods. With them we processed DrugBank and FoodBank and applied for both methods a consensus scoring approach, so in quick way, compounds in the top 1% rank for both methods were selected to visual inspection by me. Then I also checked bibliography and selected the ones of the table, first list.B) LBVS; given the tight deadline I applied a fast method called DBVS (Descriptor Based Virtual Screening). In a nutshell, using DRAGON, we calculated all descriptors for DrugBank, FoodBank and also the query compounds known to be active in coronavirus. I focused first on the one coming from the Shanghai crystal. Then I screened the compound against the two databases, and only cases in which the Euclidean distance between matching descriptors with exact same values was less than 10, then I selected them for further visual inspection and bibliography.IN this LBVS scheme we will also apply this scheme: https://chemrxiv.org/articles/Optimizing_Electrostatic_Similarity_for_Virtual_Screening_A_New_Methodology/10044272/1
Disclaimer: This following list does not take into account any pharmacological-, toxic- or side effect tested in clinical trials nor does it represent compounds directly suggested as potential drugs against 2019-nCoV. Innophore is not taking responsibility (please see our disclaimer on the top of the page).
The list we received yesterday can be downloaded from our server here: https://innophore.com/download/BIO-HPC_UCAM_list_1_of_selected_hits_submitted_to_Innophore_20200201.xlsx.
UPDATE February 5th 202013:34UTC
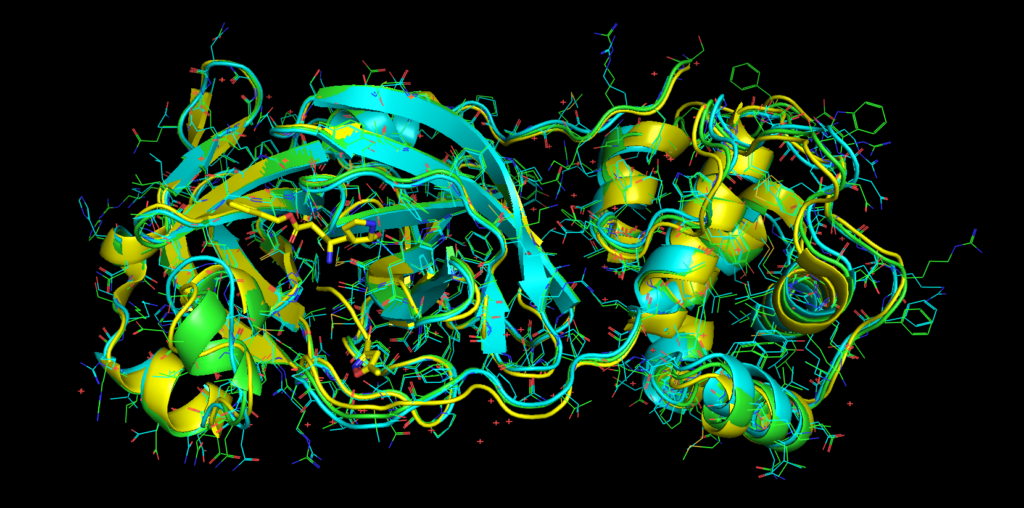
The crystal structure of the protease of our partner Prof. Yang group from ShanghaiTech is oficially released: https://www.rcsb.org/structure/6LU7. An alignment is posted below, we updated our DOI dataset containing the crystal structure: 10.6084/m9.figshare.11752752.
Yellow: X-ray, Other colours: Models
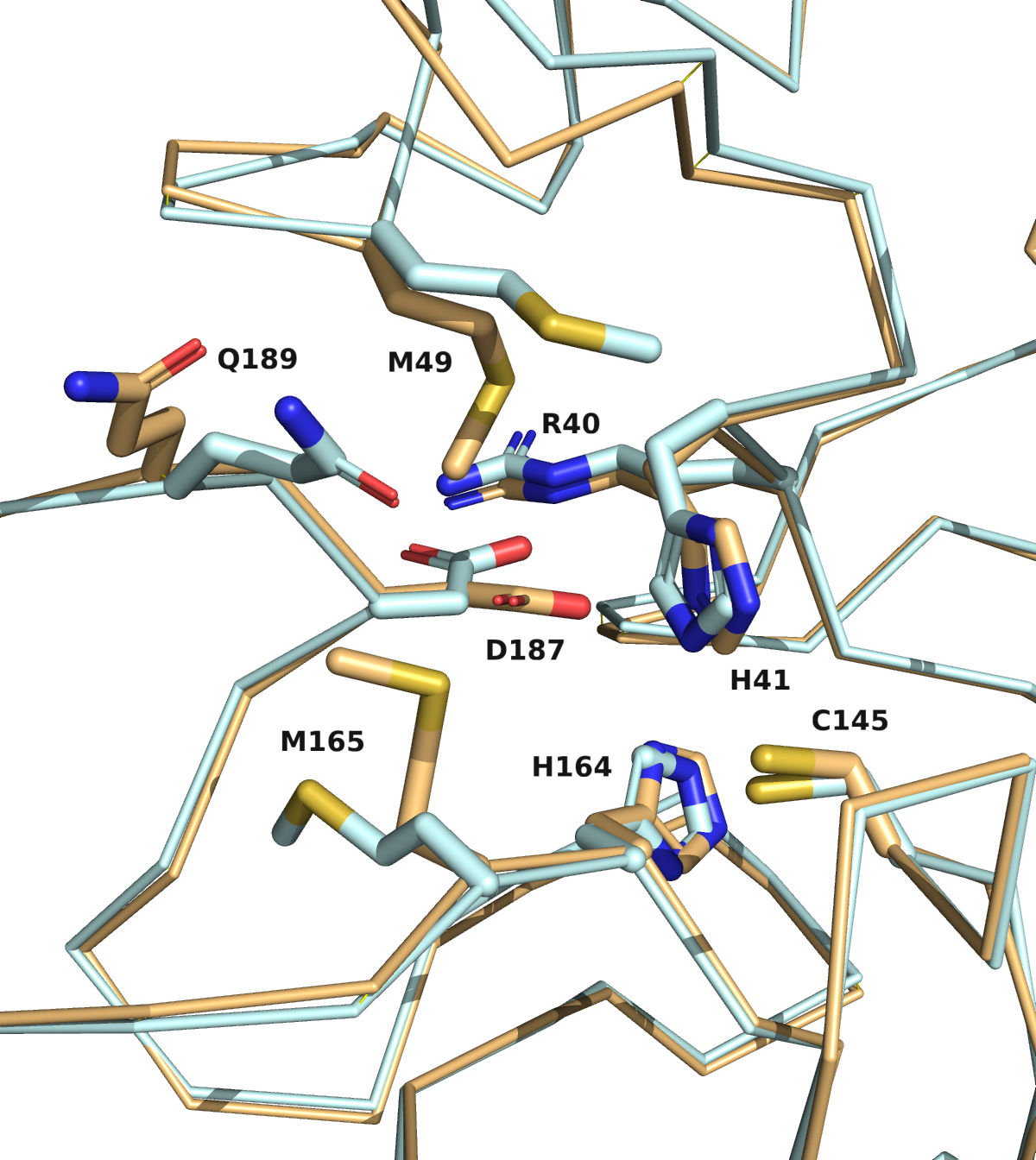
We are going to publish the MD, catalophore and docking data we produced with the crystal structure in the last 8 days very soon. Overall, the now available crystal structure of the nCov protease is very similar to the homology model previously proposed by Innophore. A Calpha-alignment resulted in an rmsd of 0.6 Angström for 282 out of 306 superimposed Calpha-atoms. As expected, several side-chains, especially on the surface of the protein, exhibit different conformations in the two structures. Such differences are also seen for residues lining the putative active site (see figure: Innophore model (orange), x-ray structure (cyan)). Particularly the differing conformations of the H41 and D187 might influence the outcome of e.g. a docking calculation if not taken into account.
UPDATE February 6th 2020 20:27UTC
We have to work together. Special situations required special efforts. It’s an incredible honour to coordinate and manage an international effort for allocating significant funding together with the University of Graz as legal coordinator to fight against the current outbreak of 2019-nCoV. In a collaborative systematic effort of in-silico and in-vitro techniques with careful balance between state-of-the art established methods fortified with emerging new technologies we aim at rapidly finding and testing chemical compounds and methods that inhibit the 2019-nCoV M(pro) protease, which is our primary target. World-wide leading institutions, active in bioinformatics, drug- screening and design supported by major European HPC facilities will be redundantly cross-linked with high-throughput experimental in-vitro and in-vivo validation methods. The consortium covers the whole pipeline, from the 2019-ncov genome isolated from patients down to the testing of compound in 2019-ncov cell cultures. Our website (currently only listing the partners) was launched yesterday: fastcure.net
UPDATE February 13th 2020 13:48UTC
Yesterday we published the next series of molecular dynamics simulations on SARS-CoV-2 (formerly 2019-nCoV) main protease (Mpro) as open data. We explicitly invite to use the data for further analysis: https://figshare.com/projects/Coronavirus_2019-nCoV/74982.
Three-fold replicates of 0.5ns molecular dynamics simulations at 310 K of the crystal structure of SARS-CoV-2 Mpro solved by our consortium partner Prof. Yang’s from ShanghaiTech in complex with potential ligand(s) in different conformations. Our consortium now has access to extensive computational resources and will continue its bioinformatic analysis.
UPDATE February 22nd 2021 08:00UTC
We haven’t updated this page in almost a year, as this was a landing page for very preliminary data. Since January 2020, many things have changed. Therefore, the way we communicate results on SARS-CoV-2 has also changed and now follows standardized rules for publishing scientific papers again. For example, we are conducting AI-based drug design and large-scale virtual screening to identify suitable therapeutics ranging from small molecules to biopharmaceuticals, as well as further structural analysis of the virus. Below are two of our recent peer-reviewed Covid 19 publications:
A multi-pronged approach targeting SARS-CoV-2 proteins using ultra-large virtual screening
iScience – Cell Press – January 2021
https://www.cell.com/iscience/fulltext/S2589-0042(20)31218-9
Serine 477 plays a crucial role in the interaction of the SARS-CoV-2 spike protein with the human receptor ACE2
Scientific Reports – Nature Group – February 2021
https://nature.com/articles/s41598-021-83761-5
This work is referenced in Wikipedia https://en.wikipedia.org/?curid=66185220#S477G/N and in a recent paper by BioNTech.
References
- [i] https://www.who.int/publications-detail/surveillance-case-definitions-for-human-infection-with-novel-coronavirus-(ncov)
- [ii] https://www.bbc.com/news/world-asia-china-51217455
- [iii] https://edition.cnn.com/2020/01/23/china/wuhan-coronavirus-update-intl-hnk/index.html
- [iv] https://www.cbc.ca/news/health/coronavirus-human-to-human-1.5433187
- [v] https://www.reuters.com/article/us-china-health-pneumonia-who-idUSKBN1ZD16J
- [vi] https://en.wikipedia.org/wiki/Novel_coronavirus_(2019-nCoV)
- [vii] https://link.springer.com/protocol/10.1007%2F978-1-4939-2438-7_1
- [viii] https://www.asiatimes.com/2020/01/article/wuhan-disease-spread-by-bats-animals-expert/
- [ix] https://www.webmd.com/lung/news/20200122/what-to-know-about-new-coronavirus-from-china
- [x] https://proteopedia.org/wiki/index.php/HIV-1_protease
- [xi] https://bmcgenomics.biomedcentral.com/articles/10.1186/1471-2164-15-S7-S5
- [xii] https://pubchem.ncbi.nlm.nih.gov/compound/Mozenavir
- [xiii] https://www.biorxiv.org/content/10.1101/2020.01.20.913368v1
- [xiv] https://www.biorxiv.org/content/10.1101/2020.01.20.913368v1
- [xv] https://www.ncbi.nlm.nih.gov/nuccore/MN908947
- [xvi] https://www.ncbi.nlm.nih.gov/nuccore/MN908947.3?location=266:13468,13468:21555
- [xvii] https://www.rcsb.org/structure/5N5O
- [xviii] https://www.rcsb.org/structure/3TLO
- [xix] https://www.rcsb.org/structure/2A5K
- [xx] http://www.sbg.bio.ic.ac.uk/~phyre2/html/page.cgi?id=index
- [xxi] http://scop.berkeley.edu/sunid=146586&ver=1.75
- [xxii] https://www.rcsb.org/structure/2DUC
- [xxiii] https://www.rcsb.org/structure/2H2Z
- [xxiv http://europepmc.org/article/med/17189639
Notice: In the current situation our highest priority is the rapid production and publication of data to make available for scientists everywhere to continue this work and no time and no resources are wasted by starting from scratch every time.We are of course double-checking our data and trying to make sure to produce high-quality calculations and simulations. However, if you find any inconsistencies or errors please contact us anytime via mail, the website, LI, social media or phone.
One Comment
Coronavirus outbreak (covid 19) explained through 3D Medical Animation – Health Site
[…] Coronavirus COVID-19 (formerly known as Wuhan coronavirus and 2019-nCoV) – what we can find ou… […]
Comments are closed!